动态建模有助于预测肠道微生物的行为
人类的肠道充满了微生物,每个微生物都在一个令人难以置信的积极和消极交流网络中相互作用。有些产生的物质可以作为其他微生物的食物,而其他产生毒素 - 抗生素 - 会杀死邻居。
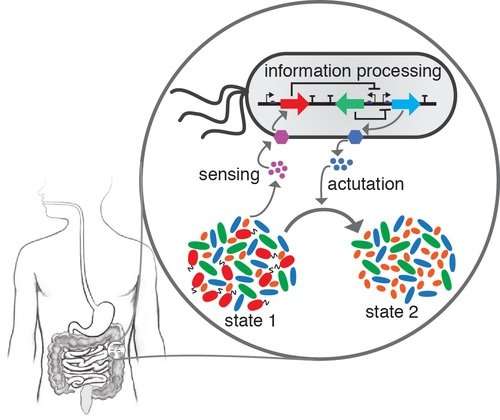
科学家一直在试图了解这种被称为微生物组的肠道微生物是如何形成的,它是如何随时间变化的,以及它如何受到用于治疗疾病的抗生素等干扰的影响。威斯康星大学麦迪逊分校的生物化学教授奥菲莉亚·文图雷利和她在加州大学伯克利分校的合作者的一项新研究可能有助于缓解一些困难。
该研究于6月21日发表在“分子系统生物学”杂志上,为预测微生物肠道群落的工作方式提供了一个平台,并代表了了解如何操纵肠道生态系统特性的第一步。例如,这可以让科学家设计一种持续存在于肠道中的益生菌,或者定制饮食以对人类健康产生积极影响。
“我们对肠道微生物组的生态相互作用知之甚少,”Venturelli说。“许多研究都集中在对所有存在的微生物进行编目,这非常有用,但我们希望尝试理解管理它们组装成社区的规则,如何实现稳定性,以及它们如何对扰动作出反应。”
通过学习这些规则,研究人员表示,他们可以使用计算工具更好地预测微生物之间的相互作用,而不是进行费力且耗时的实验室实验。
数据还可以开始回答有关病原体在入侵社区时如何造成损害的问题,以及如何预防。
在研究中,研究人员选择了人体肠道中存在的12种细菌类型。它们代表了肠道微生物组的多样性,并且大多数已显示出显着影响人类健康。它们与糖尿病,肠易激综合征,克罗恩病和结肠癌等疾病有关。
该团队收集了所谓的成对相互作用的数据,这意味着每个细菌物种只与另一个细菌物种配对,以研究两者如何相互作用,而不必担心所有其他人正在做什么。这是针对12个成员社区中每一个可能的配对而完成的。
研究人员将关于成对相互作用的数据以及每个物种的数据提供给动态模型,以解释所有细菌在组合时可能如何相互作用。他们发现仅成对数据足以预测较大社区如何组装。
“这种模式使我们能够以更少的测量结果更好地理解和预测肠道微生物组,”Venturelli解释道。“我们不需要测量每一个可能的社区,例如,一组物种中的三个,四个或五个。我们只需要测量所有对,这仍然代表一个非常大的数量,以便能够预测整个肠道的动态。“
虽然这仍然是一个挑战,但Venturelli说它将显着减少科学家需要进行的测量数量。
研究人员还通过测量微生物产生的物质(称为代谢物)来研究哪些物种似乎是社区中最重要的物种。令他们惊讶的是,“代谢物数据无法预测重要物种在社区中的作用,”Venturelli说。
然后,她和研究小组通过试图估计其12种所选细菌的不同组合的特征来测试该模型的预测能力。虽然不完美,但该模型在预测动态行为方面做得很好。
此外,研究小组发现,社区中微生物之间的相互作用比基于其他主要表现为负相互作用的研究所预期的更为积极。
“我们发现有正面和负面的相互作用和之间的平衡消极互动那种为社会提供一种稳定的力量,”勒里说。“我们开始了解肠道微生物稳定性的设计原则,以及社区从扰动中恢复的原因。”
该模型允许科学家现在开始询问有关数千个微生物群落的组成和动态的问题。
“如果没有模型,我们基本上只是盲目地测试事物,而不是真正了解我们正在做什么以及当我们做什么时,例如,试图设计干预措施,”她说。“拥有一个模型是能够以有益于人类健康的方式操纵肠道生态系统的第一步。”
推荐内容
-
心理咨询之婚恋观——如何度过七年之痒_度过七年之痒方法
面对现在的社会压力,很多人选择先发展事业再决定谈恋爱结婚这件事。毕竟很多人都觉得爱情可遇不可求,不是说你努力去脱单就能脱单的,...
-
婚后夫妻怎样互相了解_婚后夫妻怎样相处
生活中,不管我们在跟自己独处还是跟别人相处的过程中其实我们会发现心理对一个人的影响是很大的。因此当我们发现自己有心理疾病的倾向...
-
心理咨询之婚恋观——造成婚外恋的原因是什么
感情这种事谁都说不准,与其自己一个人瞎猜不如看下经验者是如何应对这些问题的,多一个参考就能避免一些不必要的误会,了解的更多就会...
-
为预防视网膜脱离 高度近视眼患者每年应做一次散瞳检查眼底
前不久,小王和朋友一起去游乐场坐过山车,玩得很尽兴。可回来后几天,他总感觉眼前有黑影飘过,还有一点一点的亮光闪烁。家人陪同他到...
-
植物中非编码RNA的功能是什么
DNA拥有生命的蓝图。然而,高等生物中的DNA很少是蛋白质编码基因。例如,在人类中,只有3%的DNA代表基因。问题是:其他97%的人做了什么?奇
-
如何解决做家务问题
现实生活中,我们很难揣测出一个人的心理活动。但是我们可以根据一些知识的细节,去假设判断一些人的心理,也包括对自己的心理的认知。 有
-
如何摆脱失恋的痛苦
日常生活中,我们会发现一个高情商的人是很受欢迎的。那么如何做一个情商高的人呢?其实只要懂得一点心理观察,善于捕捉一些细节,我们...
-
潮流健身好处有那些
【潮流健身有哪些】潮流健身好处有那些前言:如何为精神压力大的当代年轻人减压?这个问题已经迫在眉睫。除了政府和相关组织的科普,更...
-
分析如何通过各种办法提升幸福感
生活中,不管我们在跟自己独处还是跟别人相处的过程中其实我们会发现心理对一个人的影响是很大的。因此当我们发现自己有心理疾病的倾向...
-
《今夕何夕》冯夕为什么能听见笛声 冯夕另一身份是尔玉吗
新生代渐渐成为影视圈的主力军,因为在这看脸的时代,长得年轻帅气、漂亮精致的男女明星颇受观众喜爱,所以他们自然而然拥有很大的需求...



