使用metaBAT改善微生物基因组分组
DOE JGI研究人员开发了一种名为MetaBAT的自动化工具,可自动对从宏基因组序列组装的大型基因组片段进行分组,以重建单个微生物基因组。从大型复杂宏基因组数据集中准确有效地重建个体微生物基因组的能力使研究人员能够了解它们各自如何相互作用并影响全球周期。MetaBAT等工具使研究人员能够更好地了解高通量宏基因组测序产生的数据,这使得研究人员无需培养微生物群落即可对其进行研究。

如果几克土壤可以容纳多种微生物相互作用以影响全球碳循环,那么想象一下,牛的瘤胃中有多少微生物有助于分解植物质量以获取养分,这些信息可能对从植物开发可持续的替代燃料。技术进步使研究人员能够利用高通量宏基因组鸟枪测序来研究微生物群落,而无需培养这些生物。然而,研究人员仍在研究如何从这些大规模数据集中有效和准确地组装单个微生物基因组,以了解每个人对维持全球周期的具体贡献。
目前大多数从宏基因组数据集“分组”或分组大基因组片段以重建个体基因组的方法具有局限性。它们中的一些依赖于已知的基因组作为参考,这种方法在环境样品上不能很好地工作,其中许多微生物与已知的基因组没有密切相关的物种。其中许多包含手动步骤,并且无法扩展以处理大型宏基因组数据集。
在PeerJ于2015年8月27日发表的一篇论文中,来自美国能源部联合基因组研究所(DOE JGI)的研究人员,DOE科学用户设施办公室,提供了一种自动化的宏基因组装箱软件工具,可以解决这些障碍。描述MetaBAT(用于具有丰度和四核苷酸频率的Metagenome Binning)的论文被选为代表“PeerJ在2015年10月之前的2年中发表的一些最值得注意的基因组学研究”的10篇文章。
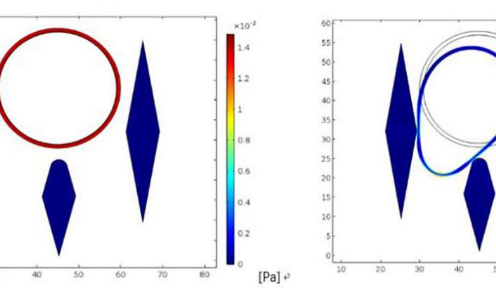
该团队使用合成和真实世界宏基因组数据集评估MetaBAT,将使用该工具精确回收的基因组区域数量与其他分级方法发现的基因组区域进行比较。他们发现MetaBAT恢复了“许多[基因组]错过了替代工具。” 更重要的是,他们报告说MetaBAT具有计算效率; 该软件从合成的宏基因组数据集中识别出340个箱,仅需14分钟即可获得200,000个碎片,仅使用3.9 GB的RAM,而其他方法需要20到104个小时,并使用9.5GB到38GB的RAM来获取相同的数据。
MetaBAT是一个开源软件工具,可从https://bitbucket.org/berkeleylab/metabat获得,并附有软件手册。该工具已被自然研究中的研究人员用于应用组学来研究永久冻土中的微生物群落,以及在一项科学研究中报告重建来自古菌门的深层地下水产甲烷菌的完整基因组。甲烷代谢。
推荐内容
-
大秦赋辛柏青张鲁一被指强行装嫩 大秦赋张鲁一第几集出场
在等电视剧大秦赋辛柏青张鲁一被指强行装嫩 大秦赋张鲁一第几集出场上映的时候,看完了大秦赋辛柏青张鲁一被指强行装嫩 大秦赋张鲁一第几
-
微小的过滤器有助于检测癌细胞
多发性骨髓瘤是一种血液癌症,其中恶性浆细胞(一种白细胞)积聚在骨髓中。这会导致骨骼破坏和骨髓衰竭,在健康个体中会产生人体所有的红...
-
《狼殿下》里的朱温是谁演的 炀国皇帝楚馗原型是谁
《狼殿下》里的朱温是谁演的 炀国皇帝楚馗原型是谁,这部剧的制作是很精致的,每个剧的镜头和画面都很细心,小编将为你带来这部剧的相关精
-
四周岁孩子有自闭症吗
都说小孩一天一个样。其实从小到大的成长过程中每个人都在不断的“进化”四周岁孩子有自闭症吗1、自闭症是影响儿童较为严重的一种病,是...
-
青春创世纪蒋哲洋喜欢钱希西吗 蒋哲洋最后和谁在一起了
青春创世纪蒋哲洋喜欢钱希西吗 蒋哲洋最后和谁在一起了又出现了很多优秀的电视剧,最近陆陆续续出现了很吸引人目光的电视剧,那么就跟随小
-
失恋了看什么电影
面对现在的社会压力,很多人选择先发展事业再决定谈恋爱结婚这件事。毕竟很多人都觉得爱情可遇不可求,不是说你努力去脱单就能脱单的,...
-
心理咨询之婚恋观——【让你们心贴心的动作】让你们心贴心的动作
现在的年轻人对于婚姻和爱情大部分是很被动的,他们觉得感情这种事随缘就好,其实一个人也可以过的很精彩。但是没经历过你又怎么知道爱...
-
为什么美国孩子那么自信
一个人的性格跟他从小成长的环境分不开。父母亲的教育尤为重要,将会影响孩子的一生。美国的小孩,不管学习好坏、长得丑俊、高矮胖瘦,...
-
喜欢自虐的都是什么心理
社会的发展越来越快,大家现在也慢慢开始关注一个人的心理健康。因为外界事物的巨大变化,很多人跟不上变化的脚步就会产生心理落差感从...
-
阴道细菌改变寨卡病毒和单纯疱疹病毒-2的性传播
根据加尔维斯顿德克萨斯大学医学分会的一项新研究,阴道中的细菌可以抑制女性中性传播的寨卡病毒和单纯疱疹病毒-2。2017年6月2日在新奥尔良